The production stages that most increase validation risk in medical injection molding include material selection, tooling decisions, process development, process qualification, and post-processing operations, because each of these stages defines constraints that later validation activities must confirm under real manufacturing conditions.
In this context, validation risk refers to the likelihood that a process cannot consistently produce compliant medical components across different batches, environments, and timeframes, even if initial samples meet specifications, especially in complex environments such as medical device contract manufacturing and early-stage product development.
Why Validation Risk Propagates Across Production Stages
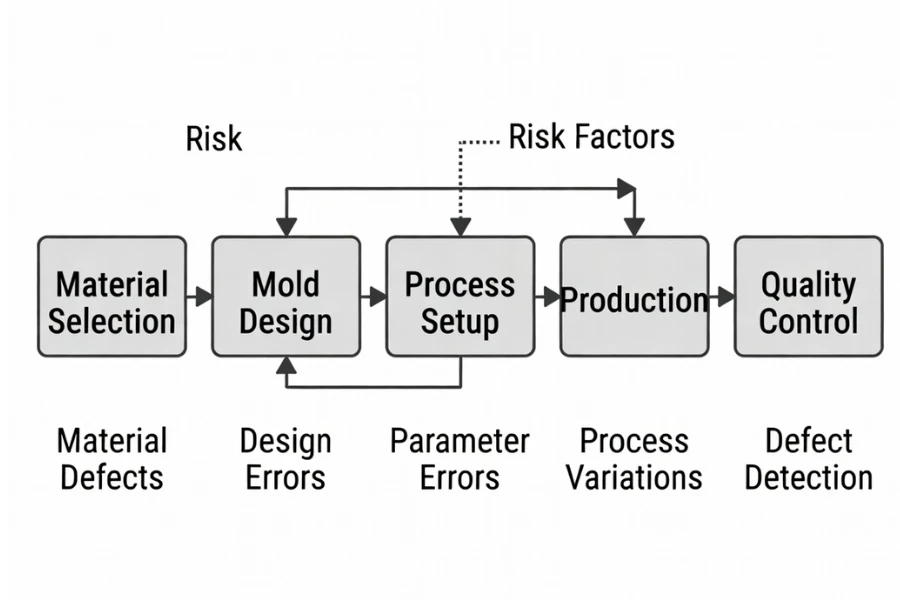
Validation challenges in medical injection molding rarely originate from a single isolated decision, because early-stage assumptions about materials, geometry, or processing conditions often become fixed through tooling and documentation, which then forces downstream qualification activities to validate conditions that may no longer reflect real-world variability.
For example, when a polymer is selected without fully verifying its behavior after sterilization, the resulting degradation may only become visible during performance testing or aging studies, while mold designs that do not adequately control venting or cooling can introduce residual stress or gas entrapment that compromises both dimensional stability and long-term reliability.
This propagation mechanism means that validation risk is cumulative rather than additive, and that the stages which define irreversible constraints tend to represent the highest exposure.
Validation Risk Mapping Across Injection Molding Stages
While all production steps contribute to overall compliance, certain stages consistently carry higher validation exposure due to their impact on process stability and downstream verification requirements.
Material selection and tooling decisions are generally considered very high-risk stages because they establish the physical and chemical boundaries of the product, whereas process development and process qualification represent high-risk stages due to their influence on repeatability and statistical capability, and downstream activities such as post-processing or lifecycle monitoring tend to represent moderate or conditional risk depending on how well earlier stages have been controlled.
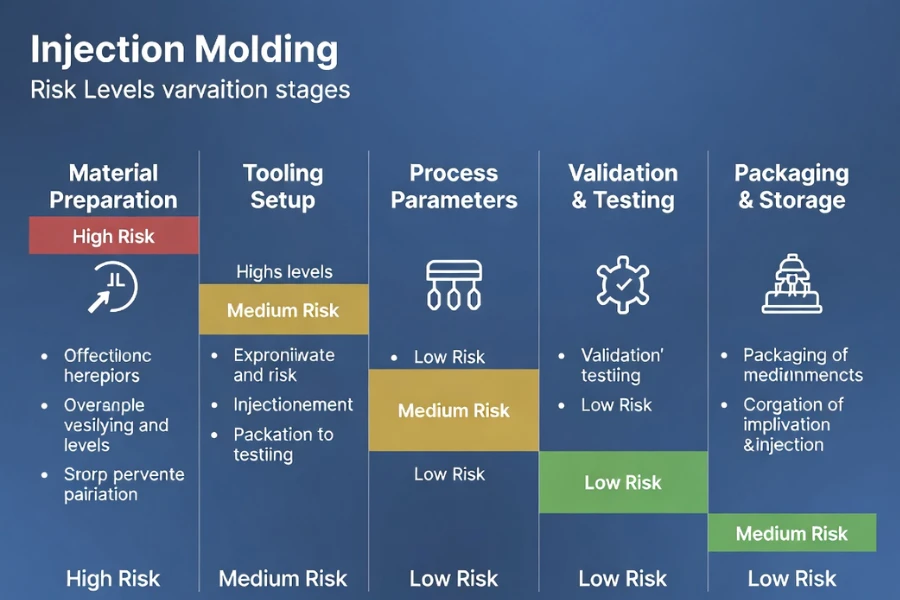
High-Risk Stages in Medical Injection Molding Production
Material Selection and Biocompatibility Assessment — Very High Risk

Material selection introduces the highest level of validation risk because it defines the fundamental behavior of the component under manufacturing, sterilization, and end-use conditions, and once a material is integrated into tooling and documentation, replacing it typically requires partial or full repetition of qualification activities.
Risk increases when ISO 10993 biocompatibility testing is not completed before mold development begins, when compatibility with sterilization methods such as ethylene oxide, gamma irradiation, or steam is assumed rather than experimentally confirmed, and when batch-to-batch variation is not evaluated under real processing conditions, since differences in melt flow index or additive content can alter shear response and affect process capability.
Another critical factor is the discrepancy between supplier-provided material data and actual molded part performance, because polymer behavior is influenced by shear rate, thermal history, and cooling conditions during injection molding, which means that validation based solely on raw material data does not guarantee compliance after processing, especially when working with different systems such as TPE materials, PP resins, or LSR silicone molding.
In stable development programs, medical-grade materials are typically locked before Alpha prototyping and validated through structured material selection processes that consider full lifecycle performance rather than isolated material properties.
Mold Design and Tooling Decisions — Very High Risk
Tooling decisions represent a very high validation risk because they physically lock geometry, tolerances, and process behavior, making subsequent design changes both costly and validation-intensive.
This risk becomes particularly significant when Class-A production tooling is introduced before critical-to-quality features are fully defined, since any modification to geometry or tolerance requires mold rework and may invalidate previously completed OQ or PQ activities, often extending project timelines by several months.
In addition to design freeze timing, mold performance itself plays a central role in validation outcomes, as insufficient control of tolerances can lead to dimensional variation that fails capability requirements, while poor gate design or inadequate venting can result in defects such as flash, short shots, or gas entrapment, all of which are unacceptable in medical components due to contamination and safety concerns.
Residual stress generated by uneven cooling or improper shrinkage compensation is another key contributor, as it may not be visible during initial inspection but can lead to cracking after sterilization or during aging, which directly impacts validation results.
To reduce exposure, early-stage programs often rely on prototype tooling or feasibility-driven mold design and manufacturing support along with early-stage tooling design optimization before committing to final production molds.
Process Development and Operational Qualification — High Risk
Process development defines the operational window within which consistent production can be achieved, and inadequate characterization of this window is one of the most common sources of validation failure during operational qualification.
Risk is elevated when validation focuses only on nominal processing parameters without intentionally challenging boundary conditions such as variations in melt temperature, mold temperature, injection pressure, environmental humidity, or production shifts, because these factors directly influence polymer flow, cooling behavior, and dimensional stability.
Thermal control is particularly critical, as fluctuations in temperature can alter viscosity and shrinkage, which affects both surface quality and internal stress distribution, and without closed-loop monitoring or sufficient experimental design, these variations can lead to a significant proportion of nonconforming parts during routine production in real plastic injection molding environments.
Additional factors such as material residence time, especially during machine stoppages or restart conditions, can cause polymer degradation that is not captured during standard validation runs but becomes evident in real manufacturing scenarios.
Robust OQ activities therefore focus on defining the full process window through structured experimentation, ensuring that acceptable product quality is maintained not only under ideal conditions but also under worst-case scenarios, particularly in regulated medical plastic injection molding applications.
Process Qualification and Capability Evaluation — High Risk
Process qualification, including IQ, OQ, and PQ, is intended to confirm that equipment, parameters, and outputs meet defined requirements, yet validation risk remains high when these activities do not fully reflect production variability or are not supported by sufficient statistical evidence.
A common issue arises when verification activities are mistaken for validation, meaning that manufacturers confirm that individual parts meet specifications without demonstrating that the process can consistently produce compliant parts across multiple batches and operating conditions.
Risk increases when PQ is conducted using a limited number of samples or a single production batch, as this does not capture variation over time, and when process capability indicators such as Cpk fall below acceptable thresholds without triggering corrective action or process adjustment.
Documentation gaps also contribute to validation exposure, since incomplete protocols or missing reports can lead to regulatory noncompliance even if the physical process performs adequately.
Effective qualification frameworks typically rely on structured medical device verification and validation (V&V) systems that align process capability with regulatory expectations.
Quality Control and Inspection Strategy — Moderate Risk
Quality control introduces moderate validation risk because, while it supports process monitoring, it cannot compensate for an unstable or poorly validated process.
Risk increases when inspection strategies rely heavily on sampling methods without considering process capability, or when critical features are not continuously monitored through automated systems, which can allow defects to pass undetected, especially in high-volume OEM medical component production environments.
Manual inspection is particularly limited in identifying subtle defects such as micro flash or particulate contamination, where consistency and detection sensitivity are critical, and over-reliance on human inspection can introduce variability that affects overall quality assurance.
A more effective approach aligns inspection intensity with process stability, increasing monitoring during early validation phases and reducing it as statistical confidence is established, while maintaining full inspection for high-risk characteristics that directly impact patient safety.
Post-Processing, Sterilization Compatibility, and Packaging — Moderate Risk
Post-processing and packaging activities are often underestimated in validation planning, yet they directly affect product safety, contamination control, and regulatory compliance, especially in medical environments where cleanliness and sterility are critical.
Residual burrs from trimming operations can become sources of particulate contamination if not properly controlled, while sterilization processes can alter material properties, requiring validation of mechanical performance, dimensional stability, and biocompatibility after sterilization and aging rather than before.
Packaging systems must also be validated to ensure integrity and contamination prevention during storage and transportation, which involves simulating real-world conditions rather than relying on static tests, and these processes are often integrated within broader secondary operations such as medical device assembly and medical device packaging.
Manufacturing in controlled environments, such as ISO 7 or ISO 8 cleanrooms, helps reduce environmental variability and contamination risk, and production systems such as cleanroom injection molding improve overall validation consistency.
At SeaSkyMedical, cleanroom-based manufacturing and integrated secondary operations are structured to align downstream processes with earlier validation requirements, which helps reduce late-stage compliance risks without introducing unnecessary process complexity.
Revalidation After Production Changes — Conditional High Risk
Production changes introduce conditional high validation risk because even minor modifications can alter process behavior or product performance, requiring reassessment of previously validated conditions.
Risk arises when changes such as material supplier substitution, tooling replacement, equipment relocation, or process parameter adjustments are implemented without a structured impact assessment, since these changes can shift the process window and invalidate existing validation data.
Even seemingly minor differences, such as variation in melt flow index between material batches or wear in mold components, can affect repeatability and dimensional stability, which may not be immediately visible but can lead to long-term compliance issues.
Effective change control systems evaluate the impact of each modification and define corresponding revalidation requirements, ensuring that validation remains aligned with actual production conditions.
Lifecycle Monitoring and Continuous Improvement — Moderate Risk
Lifecycle monitoring represents a moderate validation risk because it determines whether validation remains effective over time, rather than at a single point during product launch.
Risk increases when manufacturers do not collect or analyze production data and customer feedback, which limits their ability to detect trends such as declining process capability or emerging defect patterns that were not identified during initial validation.
Over time, process drift caused by equipment wear, environmental variation, or material differences can reduce capability, and without continuous monitoring, these changes may not be addressed until they result in nonconformance or product failure.
Closed-loop systems that integrate corrective and preventive actions help maintain long-term stability by feeding real-world data back into validation planning and process optimization.
How to Reduce Validation Risk in Practice
Reducing validation risk in medical injection molding requires aligning early-stage decisions with downstream requirements, which means selecting materials based on full lifecycle performance rather than initial data, delaying hard tooling until design stability is confirmed, and defining process windows through comprehensive experimentation that reflects real production variability.
It also requires integrating process qualification with statistical monitoring, ensuring that inspection supports rather than replaces validation, and maintaining structured change control and lifecycle monitoring systems that keep validation aligned with actual manufacturing conditions.
For programs that require coordinated support across design, tooling, molding, and cleanroom production, working with an experienced partner offering custom plastic molding solutions and integrated medical device manufacturing services can help manage validation risk more effectively across all stages.
For more information, contact SeaSkyMedical to explore how structured manufacturing and validation support can improve process stability and compliance outcomes.


