Cleaning and sterilization testing for medical parts typically includes TOC, bioburden, endotoxin testing, particle analysis, cytotoxicity, and biological indicator-based sterilization validation, all of which are used to demonstrate that contamination risks are controlled and that the product can reliably achieve a sterility assurance level of 10⁻⁶.
Overview – Purpose and Core Concepts of Testing
The primary objective of cleaning and sterilization testing is to confirm that medical parts meet a sterility assurance level of 10⁻⁶, meaning the probability of a viable microorganism remaining is extremely low and acceptable for clinical use.
Cleaning is a prerequisite for sterilization because residual oils, proteins, or particles can shield microorganisms from sterilizing agents, reduce penetration efficiency, and lead to validation failure even when sterilization parameters appear correct.
Testing activities are typically divided between manufacturing and clinical use stages, where manufacturers are responsible for validating cleanliness and sterilization before product release within a medical device contract manufacturing environment, while healthcare facilities focus on verifying cleaning effectiveness and monitoring sterilization consistency during reuse.
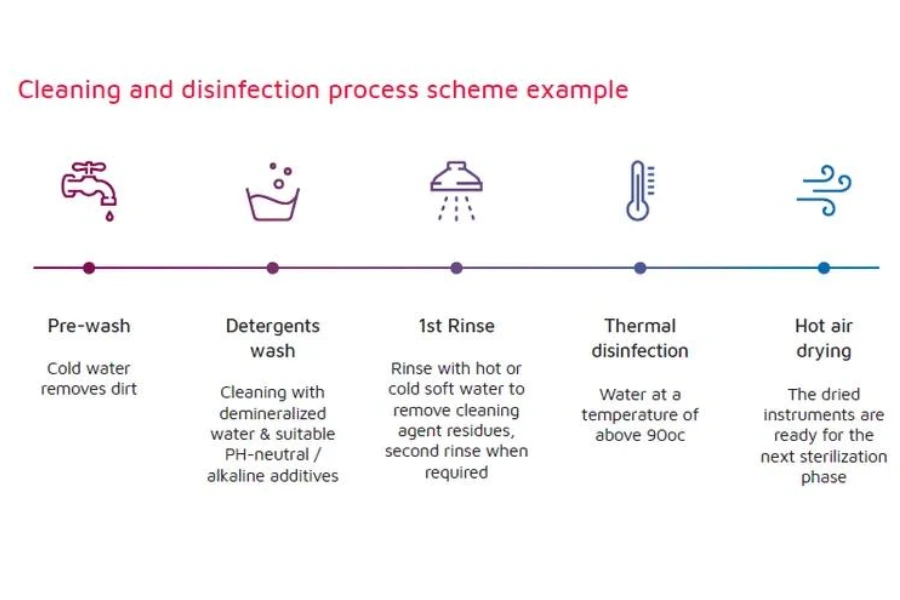
Cleaning Validation After Manufacturing
Cleaning validation ensures that residues introduced during medical plastic injection molding, CNC plastic machining, or medical device assembly are removed to levels that do not interfere with sterilization or pose risks to patients.
In medical injection molding environments, residues may include mold release agents introduced during mold making and injection processes, machining oils, polishing compounds, packaging particles, and cleaning agent remnants, all of which must be controlled through validated cleaning processes.

Chemical Residue Testing
Chemical residue testing focuses on identifying and quantifying organic contamination that may remain after cleaning, with total organic carbon (TOC) used as a broad indicator of residual organic substances such as surfactants or polishing compounds, while total hydrocarbon content (THC) provides more specific detection of oil-based residues commonly introduced during machining or lubrication processes.
In practice, TOC and THC are often evaluated together because TOC alone cannot distinguish between different residue sources, and elevated THC values typically indicate insufficient removal of process oils or coolant residues, especially in metal or hybrid components.
Sampling is usually performed using rinse or extraction methods, and variability can arise from surface geometry, material absorption, and incomplete rinsing, which means cleaning validation must consider worst-case device configurations rather than average conditions.
Microbiological Testing
Microbiological testing during cleaning validation includes bioburden measurement, which determines the number of viable microorganisms remaining on the device prior to sterilization, and endotoxin testing using the LAL method, which detects pyrogenic substances that can trigger inflammatory responses even after sterilization.
Bioburden data is not only a cleanliness indicator but also a critical input for radiation sterilization validation, where sterilization dose selection depends on the initial microbial load present on the product.
Particle and Toxicity Testing
Particle analysis is used to identify residual contaminants such as metal fragments, fibers, or environmental particulates that may originate from molding, trimming, or medical device packaging processes.
Cytotoxicity testing evaluates whether any remaining chemical residues can cause adverse biological responses, and it is particularly relevant for manufacturing-stage validation because it addresses risks associated with production materials rather than clinical contamination.
Compared with reprocessing validation, cytotoxicity testing is unique to manufacturing because it reflects the need to ensure that devices are safe before they are ever exposed to patients.
Sterilization Validation After Manufacturing
Sterilization validation demonstrates that a defined process can consistently achieve SAL 10⁻⁶ for a specific device design, material combination, and packaging configuration, and it must account for worst-case conditions such as maximum load density and the most difficult-to-sterilize locations.
Steam Sterilization Validation
Steam sterilization validation uses biological indicators containing Geobacillus stearothermophilus spores, which are placed at locations such as internal joints, dense packaging areas, or device lumens where steam penetration is most challenging.
Validation typically follows a half-cycle approach in which exposure time is reduced, and all biological indicators must remain inactive across multiple consecutive cycles before the process is considered acceptable.
Supporting tests such as Bowie-Dick testing and vacuum leak testing ensure that air removal and chamber integrity do not compromise sterilization performance, particularly in pre-vacuum systems.
Steam sterilization is typically applied to heat-stable and moisture-resistant materials, and failure often occurs when trapped air pockets or excessive load density prevent uniform steam contact.
Radiation Sterilization Validation
Radiation sterilization validation is based on establishing a relationship between product bioburden and radiation dose, rather than relying on fixed biological indicator challenges, which makes accurate bioburden measurement essential.
A verification dose is applied to product samples to demonstrate a reduced sterility assurance level, and successful sterility results support the use of a higher routine dose capable of achieving SAL 10⁻⁶.
Dose mapping is required to confirm that all parts of the load receive sufficient radiation, and failure risks are often associated with uneven density distribution or shielding effects within packaged products.
Radiation sterilization is commonly used for high-volume disposable medical components where consistent material compatibility and throughput are required.
Ethylene Oxide (EO) Sterilization Validation
EO sterilization validation uses Bacillus atrophaeus spores placed in the most difficult-to-penetrate areas, such as long, narrow lumens or multi-channel devices, where gas diffusion is inherently limited.
The process is controlled by interdependent parameters including gas concentration, temperature, humidity, and exposure time, and deviations in any of these variables can significantly affect sterilization outcomes.
Residual testing is required to ensure that EO and its byproducts are reduced to safe levels, as materials can absorb gas during sterilization and release it over time.
EO sterilization is typically selected for complex, heat-sensitive devices where other methods cannot achieve adequate penetration without damaging materials.
Other Sterilization Methods
Other sterilization methods such as vaporized hydrogen peroxide and dry heat follow similar validation principles, with biological indicators used to confirm microbial lethality and additional considerations given to material compatibility and penetration characteristics.
Dry heat sterilization may also include depyrogenation validation to demonstrate the removal of endotoxins, which is critical for certain high-risk applications.
Reprocessing Validation for Reusable Medical Devices
Reprocessing validation ensures that reusable devices can be effectively cleaned and sterilized after clinical use, with emphasis on removing biological contamination such as blood, tissue, and microbial residues.
Cleaning Effectiveness Testing
Cleaning effectiveness is evaluated through protein residue testing, often using colorimetric methods, as well as TOC analysis to detect organic contamination and bioburden testing to quantify remaining microorganisms.
Rapid methods such as ATP bioluminescence provide immediate feedback but are typically used as screening tools rather than definitive validation methods.
Compared with manufacturing cleaning validation, these tests focus on biological contamination rather than production residues, reflecting the different risk profile of used devices.
Sterilization Process Monitoring
Sterilization monitoring in healthcare settings relies on chemical indicators to confirm exposure to sterilization conditions and biological indicators to verify microbial inactivation, with monitoring frequency determined by device criticality and usage patterns.
Mechanical monitoring of temperature, pressure, and time is performed for every cycle, providing a continuous record of process conditions that supports traceability and investigation if deviations occur.
Complex Device Challenges
Devices with complex geometries, such as endoscopes, require additional validation using lumen challenge devices and biofilm simulation models, as these structures can harbor microorganisms that are difficult to remove through standard cleaning processes.
These challenges highlight the importance of both device design and validated reprocessing instructions in ensuring patient safety.
Routine Monitoring and Quality Assurance in Sterile Processing
Routine monitoring combines mechanical, chemical, and biological methods to ensure that sterilization processes remain consistent during daily operation, with each method providing a different level of assurance.
Daily Bowie-Dick testing verifies air removal in pre-vacuum steam sterilizers, while biological indicators are used at defined intervals to detect potential sterilization failures before they impact patient care.
Additional quality checks, such as monitoring rinse water for residual cleaning agents, help ensure that upstream cleaning processes remain effective and do not introduce new risks.
Handling Positive Test Results and Process Deviations
When a positive biological indicator or other abnormal result occurs, a structured investigation is required to determine whether the result reflects a true process failure or an artifact caused by testing conditions or contamination.
This may involve repeating sterilization cycles, reviewing process data, and performing additional microbiological analysis, with decisions based on a combination of biological, chemical, and mechanical evidence rather than a single test result.
For implantable devices, a more conservative approach is typically taken, and products are not released until sterility has been confirmed through validated testing.
Design Considerations That Influence Testing Outcomes
Cleaning and sterilization performance is strongly influenced by product design, as features such as narrow lumens, complex joints, and multi-material assemblies can significantly increase the difficulty of residue removal and sterilant penetration.
Surface roughness, material absorption characteristics, and packaging configuration all affect validation outcomes, and failure to consider these factors early in development can lead to repeated validation failures or costly redesigns.
For this reason, design-for-manufacturing and design-for-sterilization approaches are commonly applied during medical product development, where early-stage mold design optimization and proper material selection are aligned with downstream validation requirements.
Advanced processes such as micro injection molding and medical insert molding further increase design complexity, requiring careful consideration of cleaning accessibility and sterilization penetration paths.
SeaSkyMedical integrates these considerations into its ISO-controlled cleanroom injection molding environment, supporting consistent cleaning performance, stable bioburden levels, and predictable sterilization validation outcomes across a wide range of OEM medical components.
Conclusion
Cleaning and sterilization testing for medical parts forms a coordinated system that controls contamination risks from manufacturing through clinical use, where cleaning validation addresses production residues, sterilization validation ensures microbial inactivation, and ongoing monitoring maintains process consistency.
By understanding not only what tests are performed but also how design, materials, and manufacturing conditions influence their results, manufacturers can develop medical components that meet regulatory expectations while ensuring reliable performance in real-world healthcare environments.